Virtual Screening of a Series of Phytocompounds from Polygonum cuspidatum for Identification of Potential Antibacterial Drug Candidates: an In-silico and Drug Design Approaches
Abstract
In recent times, the emergence of Clostridium perfringens has posed a significant challenge to public health due to its antibiotic resistance and the formation of biofilms. It is the neuraminidase enzyme that supplies toxin secretion from C. perfringens. Since the sialic acid bond is a target recognition point for bacteria, new molecules are needed to treat infections caused by dangerous pathogens such as C. perfringens. The present work focused on an alternative strategy using compounds from Polygonum cuspidatum Sieb. et Zucc. Nine bioactive compounds derived from this plant emodin, physcion, emodin-1-O-β-D-glucopyranoside, emodin-8-O-β-D-glucopyranoside, physcion-8-O-β-D-glucopyranoside, 2-methoxy-6-acetyl-7-methyl juglone, torachrysone-8-O-β-D-glucoside, polydatin and resveratrol were used as ligands and coupled. The neuraminidase enzyme from C. perfringens was chosen as the target protein. The optimal ligand insertion score and ADMET parameters were determined by employing the Lipinski rules as selection criteria. Emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside exhibited drug-like characteristics in their ability to inhibit neuraminidase, as evidenced by a chelation score of −11.9. A comparison was conducted between emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside, and the positive control quercetin. A comprehensive analysis of the drug-like properties of emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside revealed that exhibited superiority over quercetin across multiple aspects. Quercetin showed a binding affinity of −9.9, while emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside showed a binding affinity of −11.9. The results showed acceptable differential kinetic properties of emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside compared to quercetin. It has been shown to inhibit the neuraminidase enzyme from C. perfringens.
Keywords:
Pharmacokinetics, Multidrug resistance, Molecular docking, Polygonum cuspidatum, Clostridium perfringens, QuercetinIntroduction
The incidence of bacterial diseases is steadily rising, primarily due to the bacterial strains’ diminishing efficacy resulting from their evolving variants and resistance to currently available antibacterial and antiviral drugs. C. perfringens is a common pathogen in humans and livestock, causing wound infections, enteritis, and enterotoxemia, and can damage organs like the brain due to excess toxin production.1
Neuraminidase (EC 3.2.1.18) is a family of exo-sialidase enzymes that catalyze the hydrolysis of sialic acid residues from glycoproteins.2 The classification of exo-sialidases, as proposed by Henrissat in 1991, is based on their sequence and can be divided into three families: GH-33, GH-34, and GH-83.3 The glycoside hydrolase family 33 (GH-33) includes most bacterial and simple eukaryotic and trans sialidases.4 This enzyme is strictly specific according to the configuration of the glycosidic bond and relative to the position of this bond in the molecule. Generally, it cleaves α–2 → 3 and α–2 → 6 galactose, thereby specifically cleaving N-acetyl neuraminic acid (NeuSAc) from cell surface glycoproteins.5 To escape the host’s immune system, many pathogenic bacteria attach sialic acid fragments (sialylate) to their outer surface. Thus, bacterial neuraminidase (BNA) disrupts cellular homeostasis, leading to increased production of inflammatory cytokines and ultimately to inflammation and sepsis.6,7
It was discovered that certain microbes possess neuraminidase, an enzyme that controls various factors that are very important. For instance, the pathogenic effect that C. perfringens is capable of producing can only be achieved in the presence of neuraminidase. This is due to the fact that the sialic acid bond is a target identification point for bacteria. Without it, enzyme infection would be limited to one round of replication rare enough to cause disease.8 Thus, BNA regulates the release of toxins from intestinal infections and the substrate formation necessary for the metabolism of bacteria as an energy source for growth.
C. perfringens releases terminal sialic acid from glycans, secreting multi-modular sialidases that function as virulence factors, resulting in severe infections for humans, such as potentiation of α-toxin and hemolysis during blood transduction.9 It is also reported that bacterial neuraminidase (BNA) plays a crucial role in biofilm development and promotes mucosal infection.10 Thus, developing BNA inhibitors is critical to suppress bacterial pathogenic diseases and inflammation. At the same time, a variation of BNA allows the condition to evade human immune responses and thus requires the formulation of new inhibitors.
Polygonum cuspidatum (Polygonaceae) is a traditional Chinese herb that grows in Asia and North America. The roots of P. cuspidatum are listed as Huzhang in the Pharmacopoeia of the People’s Republic of China. Resveratrol, polydatin, quercetin, emodin, and their derivatives are the active phytochemical components of P. cuspidatum.11–12 These phytochemical components of this have undergone extensive research and are considered essential for the medical functions.13 In addition, anti-inflammatory, antioxidant,14 antiviral,15 antimicrobial,16 and neuroprotective17 effects of this plant were figured out previously.
Bacteriostatic effects were seen in previous studies with different extracts from P. cuspidatum. The methanol extract of P. cuspidatum root can significantly suppress bacterial neuraminidase activity and alleviate the host’s symptoms. In particular, the active ingredient emodin-1-O-β-D-glucopyranoside in P. cuspidatum extract showed a potent inhibitory effect on bacterial neuraminidase activity. The ethanol extract of P. cuspidatum has inhibitory effects on Bacillus subtilis, Staphylococcus aureus, and Pseudomonas aeruginosa with minimum inhibitory concentration (MIC) values of 100, 50, and 100 μg/mL. 18,19
Materials and Methods
Ligand preparation and selection – The 3D structures of the previously reported compounds from the P. cuspidatum plant were obtained from the PubChem chemical compounds database (https://pubchem.ncbi.nlm.nih.gov/) on 8.03.2023. Subsequently, all compounds were subjected to energy optimization using the Spartan 14 software (Version 1.1.4).
Bioactivity analysis and toxicity measurement of ligands – The SwissADME database (http://www.swissadme.ch) was used to obtain the Lipinski properties of the compounds.
pkCSM – Absorption, distribution, metabolism, excretion, and toxicity of ligands were calculated using the pkCSM online software, available at https://biosig.lab.uq.edu.au/pkcsm/.
Target protein selection and primary sequence acquisition – 1.24 was obtained from the protein database (http://www.rcsb.org) on 10.03.2023 (PDB ID:5TSP). Before the docking analysis, the enzyme structure was prepared by removing excess ligands and water molecules using the BIOVIA Discovery Studio 2021 program. Polar hydrogens were added to the protein using the AutoDock vina 1.5.7 tool, and Kollman charges were figured out. The partial charge of compounds was calculated using Compute Gasteiger.
Physiochemical properties and 3D structures of proteins optimized – To predict the properties of the selected protein target, ProtParam (https://web.expasy.org/protparam/) was used (Accessed on 10.03.2023). The number of positively charged residues (Arginine andLysine) and negatively charged residues (Aspartate and Glutamine), theoretical pI, molecular weight, ext-coefficient (including Cysteine),, instability index, aliphatic index and grand average hydrophobicity20 were calculated.
Structural analysis and functional area identification – Structural analysis BIOVIA Discovery Studio 2021 program was used.22
Active site identification – Amino acids are crucial in forming ligand-protein complexes, with protein binding pockets determined through literature searches and the CASTp database http://sts.bioe.uic.edu/castp/ (accessed 8.03. 2023).23
Molecular docking of targeted proteins – AutoDock Vina (1.5.7 tool) was used to evaluate docking and binding affinities. The best ligands were characterized according to their binding affinity. To visualize the embedded protein complexes and to understand in detail the interactions that contribute to the binding of ligands, LIGPLOT (version 2.2.8)24 and Discovery Studio (D.S.) Visualizer 2021 was used.22
AutoDock Vina is a docking program that clamps a partially flexible ligand to a partially flexible protein. It implements a genetic algorithm and is approximately 71% successful in identifying docked ligand binding modes, the same as experimental identification.25 In the current work, an AutoDock Vina function is used to describe van der Waals interactions and hydrogen bonding in terms of energies.
Lead compound identification – The two most active inhibitors were identified after a detailed analysis of the physicochemical and pharmacokinetic properties of the proteins and ligands and a comparison of the insertion score. The selected compounds were precursor compounds.
Reference antibacterial drug identification and selection – In this phase, we sought drugs that could be used to treat bacterial diseases. The Drug Bank (https://go.drugbank.com/ accessed 10.03.2023) database was used for drug identification due to its comprehensive analysis of medications, including their respective routes.
Reference drug and lead compound comparison – The reference antibacterial drug and the proposed lead compound were compared by comparing insertion scores and physicochemical and ADMET properties.
Results and Discussion
The sequence of the selected protein was obtained from UniProt with the accession number A 0A0H2YQR1. The protein is 694 amino acids long. A molecular docking approach followed by molecular dynamics simulations was used to block the biosynthetic pathways of this protein target. This protein is critical for C. perfringens to exert its pathogenic effect. The bacterial neuraminidase of C. perfringens is an important therapeutic target because of its pathogenic impact and role in biofilm development. The catalytic domain of neuraminidases has hydrophilic residues (an arginine cluster having three arginine residues and one catalytic aspartate residue) and a hydrophobic pocket.26 Cp Nani, a secreted exo-sialidase, is thought to be a potential drug target for the inhibition of human infections caused by C. perfringens.
The selected protein in UniProt (https://www.uniprot.org/) (5.03.2023) reached) Protparam was used using the identity name A0A0H2YQR1, and values were obtained. The ProtParam database was used for the estimation of both physical and different parameters. The chemical properties of the selected protein were figured out; these properties were used to calculate and evaluate the molecular weight, composition of amino acids, theoretical protein index value, atomic protein composition, extinction coefficient, the estimated half-life of protein instability, aliphatic index, and overall hydropathic mean. A PI more significant than 7 indicates that a protein is basic, while a PI less than 7 suggests it is acidic. The extinction coefficient is light absorption. An index below 40 shows protein stability, while an above 40 shows protein instability (Table 1).

Physiochemical properties of target protein
The protein from the protein data bank (https://www.rcsb.org/) (accessed on 03.03.2023) 5TSP encoded protein was obtained from the protein data bank at 1.24 resolution. Ligands and other atoms, missing polar hydrogens, were added. Energy minimization was done to receive stable conformation; BIOVIA Discovery Studio 2021 water molecules in the protein were cut from the structure, and bound ligand residues were removed. Polar hydrogens were added to the protein using the AutoDock vina 1.5.7 tool, and Kollman charges were figured out. The 3D structure of the protein was obtained from (https://alphafold.ebi.ac.uk/) (provided on 5.003.2023).
From the Interpro database (https://www.ebi.ac.uk/interpro) (accessed 03/2023). Protein has 694 amino acids. The C-terminal region is between 17-24 amino acids. The hydrophobic part of the signal peptide is between amino acids 5-16. The N-terminal region of a signal peptide is between amino acids 1-4. The 3D structure of the protein was obtained from (https://alphafold.ebi.ac.uk/) (provided on 5.003.2023).
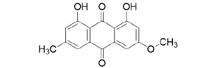
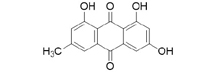
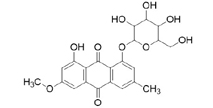
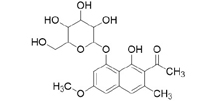
3D structures of compounds isolated from the P. cuspidatum plant were collected from the PubChem chemical compounds database (http://pubchem.ncbi.nlm.nih.gov). Selected compounds: emodin, physcion, emodin-1-O-β-D-glucopyranoside, emodin-8-O-β-D-glucopyranoside, physcion-8-O-β-D-glucopyranoside, 2-methoxy-6-acetyl-7-methyljuglone was figured out as torachrysone-8-O-β-D-glucoside, polydatin and resveratrol.
A suitable crystallographic structure of the neuraminidase enzyme (PDB ID:5TSP) from C. perfringens with a resolution of 1.24 was obtained from the protein database (http://www.rcsb.org). The SwissADME database (http://www.swissadme.ch/index.php) was used to obtain the Lipinski properties of the compounds. Table 2 shows the molecular weight, molecular formula, and 2D structure of ligands from Pubchem.
Structures of ligands with molecular formulas and molecular weights
This study used Autodock vina tools (version 1.5.7) to investigate the molecular interaction between neuraminidase enzyme and selected ligands. Before the docking analysis, the enzyme structure was optimized by removing excess ligands and water molecules using the BIOVIA Discovery Studio 2021 program. Then, all compounds were optimized for energy using the Spartan 14 (Version 1.1.4) program.
Polar hydrogens were added to the protein using the AutoDock vina 1.5.7 tool, and Kollman charges were figured out. The partial charge of compounds was calculated using Compute Gasteiger. To enable the protein to bind to its catalytic site, the x, y, and z coordinates were figured out), and the angstrom was set to 1.000. Finally, the Discovery Studio visualizer and Ligplot Molecular interactions and binding types between selected compounds and neuraminidase enzymes were investigated using (version 2.2.8) programs.27
(http://sts.bioe.uic.edu/castp/index.html?5tsp) (accessed 8.03.2023) was used to identify active sites of proteins. This software estimates available pockets for binding and gives an idea of the surface area and volume of the pockets. The active sites of the enzyme are shown in red in Fig. 1.

The 3D structural image of the selected protein from AlphaFold (A). Binding pockets of target proteins of C. perfringens neuraminidase (B).
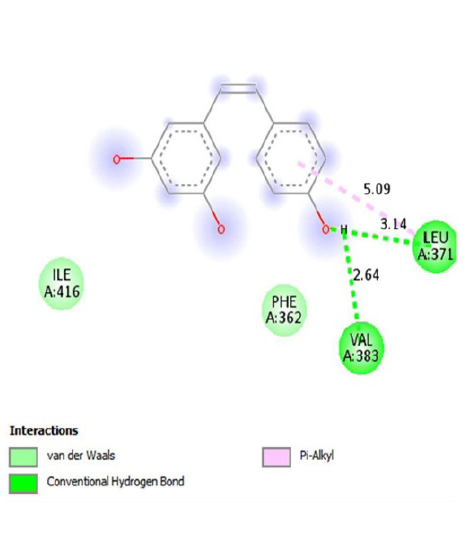
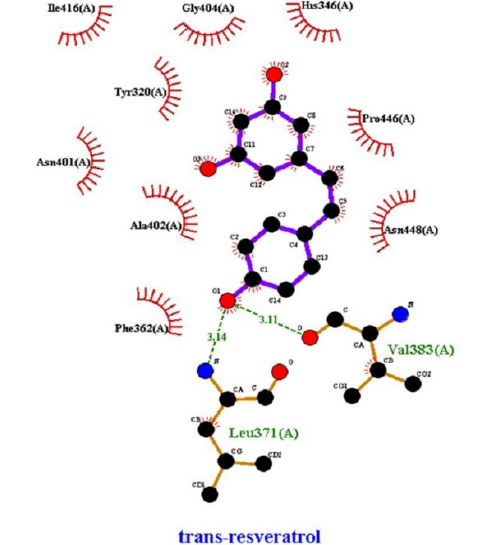
The interactions of ligands and functional pockets of proteins were calculated. To interpret the docking results, the molecular interactions and binding types between the selected compounds and the neuraminidase enzyme were investigated using the Hydrogen bond and hydrophobic bond interactions, Discovery Studio visualizer, and Ligplot version 2.2. (Table 3). Overall our results showed the bonding strengths between the residues and atoms of the ligands. The results obtained by the molecular coupling test of the molecular interaction between the selected compound and the neuraminidase enzyme are given in Table 1. The binding energy, hydrogen, and hydrophobic interactions between the compounds and the enzyme are also presented in Table 4.
Interactions of ligands with target protein.
The binding energy, hydrophobic, and hydrogen bonds of the compounds derived from the P. cuspidatum plant with the enzyme neuraminidase.
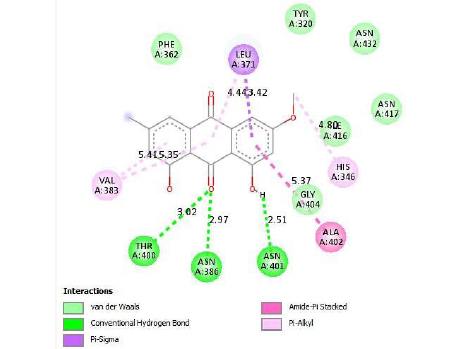
In addition, according to the results of Molecular Docking analysis, the binding energies of the studied compounds are different, therefore, the amount of binding energy ranges from −11.9 to −8.1 kcal/mol. The lower the binding energy level (negative), the stronger the binding between the receptor (enzyme) and ligands (compound or inhibitor). Among the selected P. cuspidatum compounds −11.9 kcal/mol, physcion-8-O-β-D-glucopyranoside, and emodin-8-O-β-D-glucopyranoside is predicted to supply the highest inhibitory effect since it has the lowest binding energy.
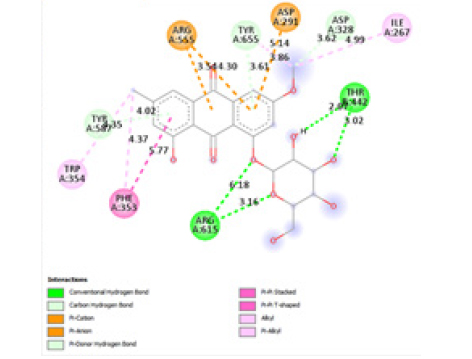
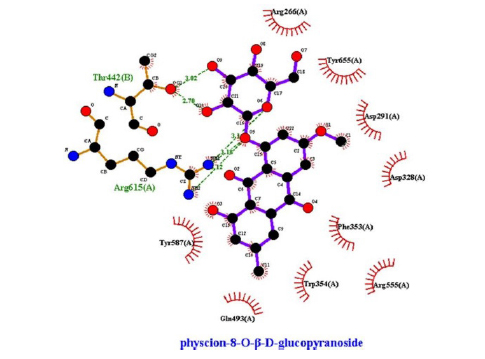
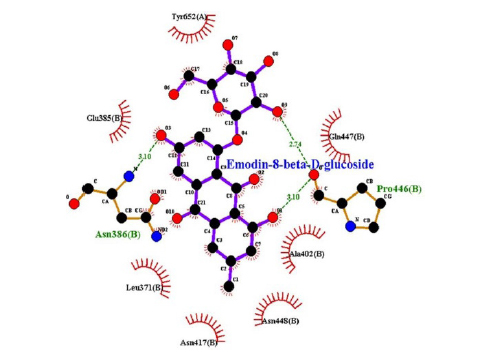
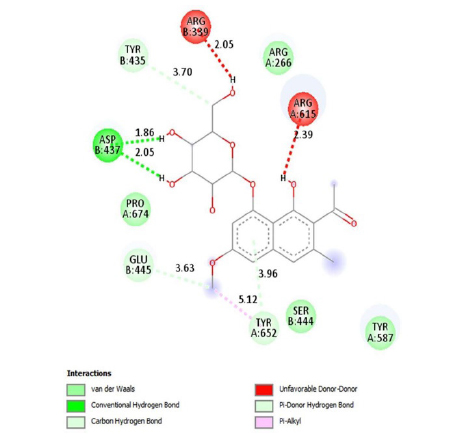
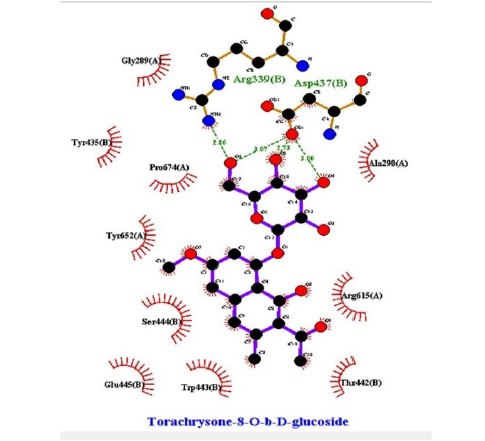
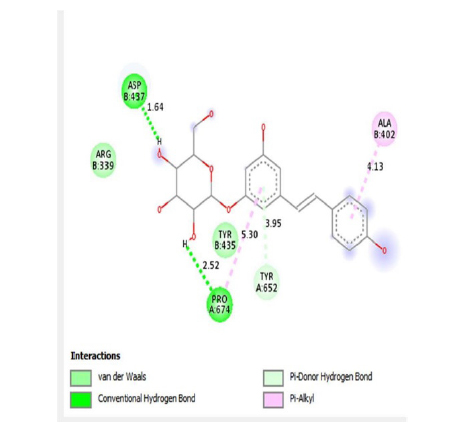
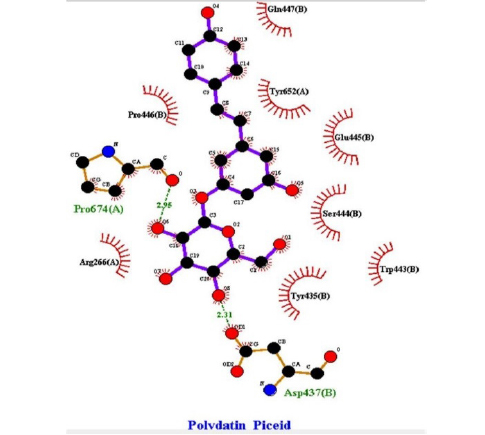
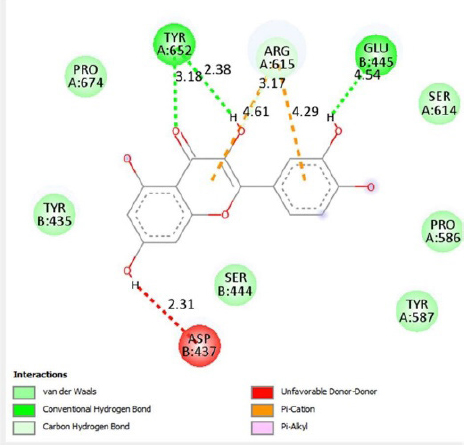
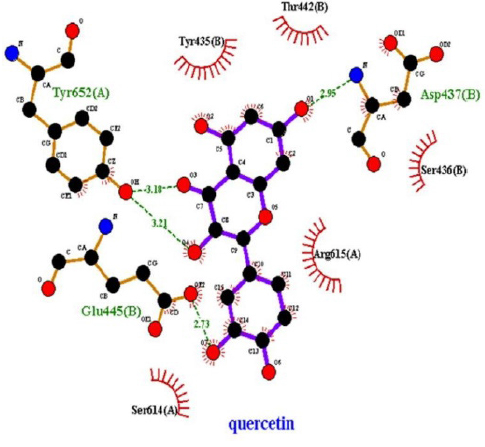
This study showed that compounds derived from P. cuspidatum can bind to the active site of the neuraminidase enzyme from C. perfringens and inhibit the activity of this enzyme. In addition, compounds named physcion-8-O-β-D-glucopyranoside and emodin-8-O-β-D-glucopyranoside, which binds with higher affinity, were detected among the isolated compounds when compared with the neuraminidase inhibitor quercetin. Hydrophobic interactions were recorded with Arg615 amino acid. The bond lengths between Tyr 652, Glu 445, and Arg615 amino acids were recorded as 2.38 , 4.54 , and 3.17 , respectively, and the bond length was recorded as 3.17 . When looking at physcion-8-O-β-D-glucopyranoside, it was seen that the number of hydrogen bonds formed with amino acids increased; these are Thr442, Arg615, Asp328, Tyr655, Tyr587 amino acids, and their bond lengths are 2.07 , 3.16 , 3.62 , 3.61 , respectively, was recorded as 4.02 . At the same time, hydrophobic interactions were seen with Tyr587, Tyr655, Phe353, Trp354, and Ile267, with bond lengths recorded as 4.02 , 3.86 , 5.77 , 4.35 , 4.99 , respectively.
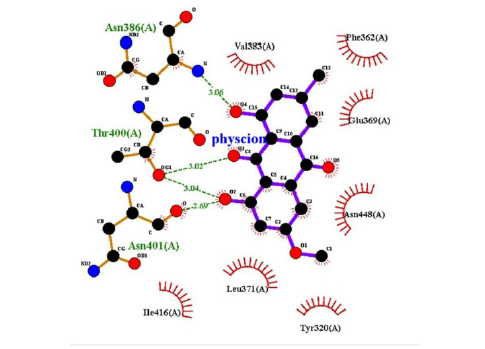
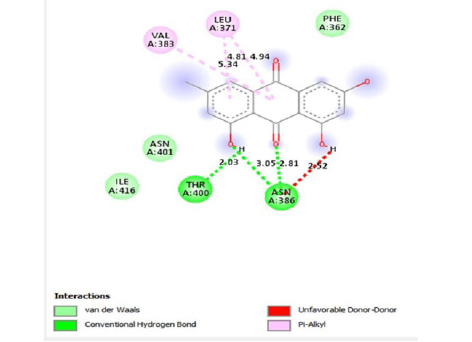
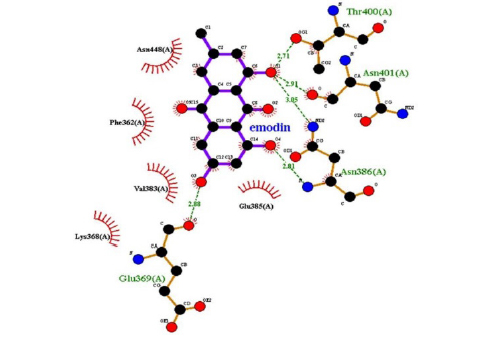
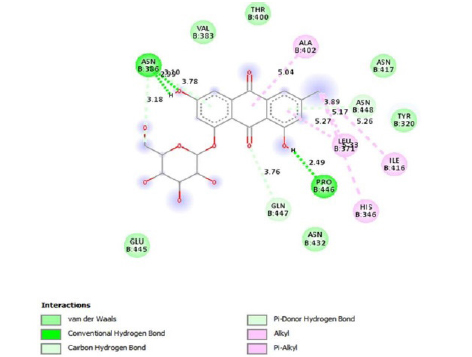
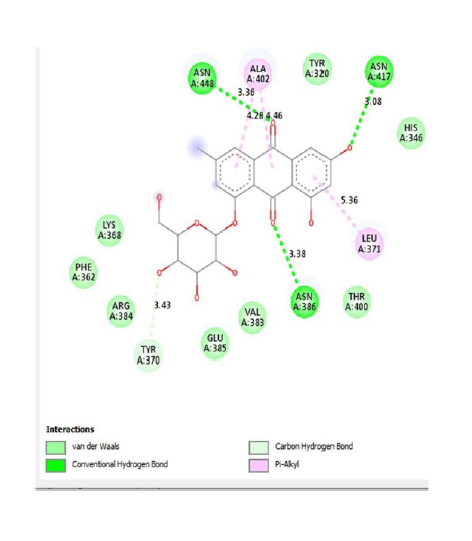
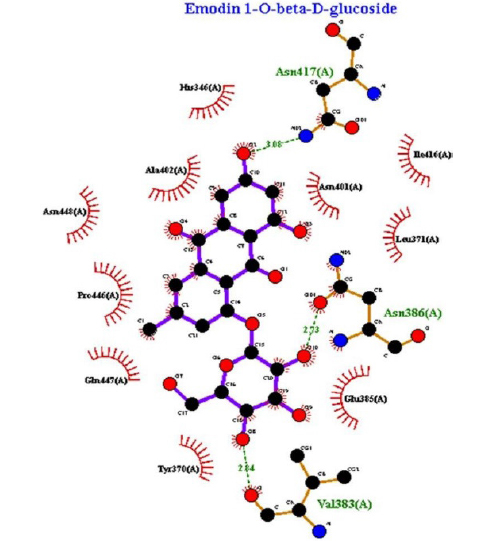
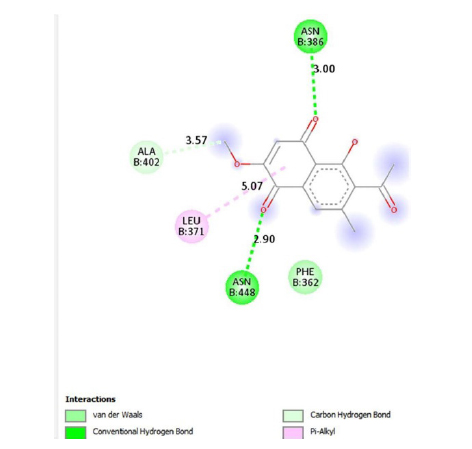
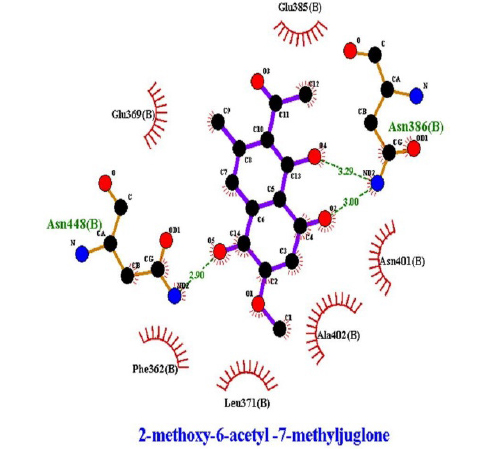
Emodin-8-O-β-D-glucopyranoside, the bond lengths between Asn386, Pro446, Asn448 and Gln447 amino acids were recorded as 2.99 , 2.49 , 3.89 and 3.76 respectively. Hydrophobic interactions were seen between Ala402, His346, Leu371, and Ile416, and the bond lengths were recorded as 5.04 , 5.33 , 5.17 , and 5.26 respectively.
In the molecular chelation analysis, it was observed that the compounds interacted with more amino acids in the catalytic region of the protein when compared to quercetin, which was used as a control, and this showed that especially physcion-8-O-β-D-glucopyranoside and emodin-8-O-β-D-glucopyranoside compounds showed interactions with the protein’s catalytic region. It shows that it is better placed in the quercetin domain, and its inhibitory effect is more effective. It is proof that it settles in the region very well, especially since the bond lengths in the emodin-8-O-β-D-glucopyranoside hydrogen bond interactions are shorter than the bond lengths in quercetin. The p compound was another vital interaction in the study; the binding affinity value was recorded as −11.2 on the docking server.
Drug similarity and ADMET properties of ligands were calculated using the online software pkCSM to qualitatively evaluate the chance of being an oral drug in terms of bioavailability and the structural or physicochemical properties of the compounds. It was found that water solubility and skin permeability were low for all ligands, and CaCO2 permeability was normal. Intestinal absorptions were more than 90% for physcion, 2-methoxy-6-acetyl-7-methyljuglone, and resveratrol ligands, while this ratio was normal for emodin and low for other ligands. All ligands show a positive P-gp substrate value except 2-methoxy-6-acetyl-7-methyl juglone, while all ligands show a negative P-gp I inhibitor value except polydatin. All ligands show negative values in the P-gp II inhibitor (Table 5).
Absorption properties of ligands
In distribution, steady-state volume of distribution (VDss), unbound fraction (Fu), blood-brain barrier permeability (Log B.B.), and central nervous system permeability (Log P.S.) parameters were examined. It was found that the VDSS values of all ligands were low, and the Fu values were positive. Log B.B. in emodin-1-O-β-D-glucopyranoside, emodin-8-O-β-D-glucopyranoside, physcion-8-O-β-D-glucopyranoside, Torachrysone-8-O-β-D-glucoside, and polydatin ligands values less than −1; log P.S. values were found to be less than −3 (Supplementary Table S1). Some isoforms of P450 enzymes involved in drug metabolism (CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4) are among the metabolic parameters. No/yes answers are given for these parameters (Supplementary Table S2). In excretion, organic cation transporter 2 (renal OCT2 substrate) was calculated with yes/no responses, while the total clearance log (CL tot) was calculated (Supplementary Table S3).
The most considerable tolerated dose amounts (MRTD) of 2-methoxy-6-acetyl-7-methyljuglone and polydatin were found to be greater than 0.477. The prime tolerable doses of these ligands are high. All ligands were found to show no inhibition of either hERGI or hERGII. None of the ligands showed hepatotoxicity and skin sensitization. In addition, no ligand showed T. pyriformis activity less than −0.5 log μg/L. Minnow toxicity values of all ligands are greater than 0.5mM, considered safe (Supplementary Table S4).
LogP values, molecular weights, hydrogen acceptor, and donor values of 9 ligands in P. cuspidatum were calculated. These results show that the compounds are poorly absorbed when there are two or more violations. Emodin-8-O-β-D-glucopyranoside, emodin-1-O-β-D-glucopyranoside, and polydatin ligands violated only one rule. All ligands obey Lipinski rules (Table 6).
Applicability of Lipinski’s rule to ligands
Binding score, ADMET properties, physicochemical properties of emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside were selected as the precursor compound capable of inhibiting target proteins according to Lipinski’s rule of five.
A comparison was made using different parameters, including ADMET properties and the physicochemical properties of compounds and positive controlquercetin, against bacterial infections due to its repeated use and efficacy, selected as the reference drug. ADMET properties include values for Drug absorption, distribution, metabolism, excretion, and toxicity. These values helped us figure out the effectiveness and efficacy of the drugs.
The absorption properties of emodin-8-O-β-D-glucopyranoside, physcion-8-O-β-D-glucopyranoside and quercetin, were compared (Table 7A).
The distribution characteristics of emodin-8-O-β-D-glucopyranoside, physcion-8-O-β-D-glucopyranoside and quercetin, were compared (Table 7B). Emodin-8-O-β-D-glucopyranoside has a higher VDss value than quercetin. Log B.B. and Log P.S. values for the two ligands were almost the same as for the control.
The metabolic properties of quercetin, physcion-8-O-β-D-glucopyranoside and emodin-8-O-β-D-glucopyranoside were compared (Table 7C). CYP1A2 and CYP3A4 inhibitors were found to be only in quercetin.
The excretion properties of emodin-8-O-β-D-glucopyranoside, physcion-8-O-β-D-glucopyranoside and quercetin, were compared (Table 7D). The total clearance value of quercetin was higher than the other two ligands, showing that it was excreted better.
The most significant tolerated dose is 1.062, 0.232, and 0.374 for quercetin, physcion-8-O-β-D-glucopyranoside, and emodin-8-O-β-D-glucopyranoside, respectively. In addition, the acute oral toxicity of the two ligands is higher than that of quercetin (Table 7E).
Quercetin, physcion-8-O-β-D-glucopyranoside, and emodin-8-O-β-D-glucopyranoside were compared according to Lipinski’s rule of five (Table 7). The logo value of quercetin and hydrogen bond donors and acceptors gave better results than the other two ligands.
Antibiotics are used as the primary source in the28–30 global management of infectious diseases. Antibiotic resistance in pathogenic bacteria is vital because it can cause a global health problem. Resistance to antibiotics has forced researchers to seek alternative approaches and new ways.31
In the current study various computational methods were used to discover a new non-toxic natural antibacterial compound for treating infectious diseases and P. cuspidatum was selected for screening in this study. This plant was chosen because it has been shown to have exceptional antipathogenic effects. The root of P. cuspidatum contains several vital phytochemical components: resveratrol, polydatin, quercetin, emodin, and their respective derivatives. Considering the plant’s therapeutic potential, the best nine ligands were selected from the studies. C. perfringens remove terminal sialic acid from glycans, thereby acting as a pathogenic effect. The pathogenic protein Neuraminidase was chosen as the target protein. The physiochemical properties, domain identification, and binding pockets of this protein were figured out by literature searches. The FASTA sequence of this protein was from UniProt, and the 3D constructs were from Alphafold. Ligands were prepared for their drug-like properties. A molecular docking protocol was applied to control the binding affinities leading to the formation of hydrogen bonds and other linkages, including hydrophobic interactions.
After a detailed analysis of ADMET properties and placement scores, the two top-rated compounds, physcion-8-O-β-D-glucopyranoside and emodin-8-O-β-D-glucopyranoside, were selected. The two chosen compounds were identified as lead compounds based on their binding affinity to the neuraminidase target protein. According to the literature, emodin-1-O-β-D-glucopyranoside strongly inhibited bacterial neuraminidase activity at a low concentration (IC50 = 0.43 μmol/L).18 Anti-inflammatory, antioxidant,14 antiviral,15 antimicrobial,16 and neuroprotective17 effects of P. cupidatum were figured out. More research is needed to clarify the exact mechanisms of action and determine its effect and safety on the human body. Neuraminidase enzyme is primarily responsible for the pathological effect of C. perfringens. C. perfringens can exert its pathogenic effect only in the presence of neuraminidase because the sialic acid bond is a target recognition point for bacteria.
Without this enzyme activity, the bacteria cannot secrete toxic cytokines. If emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside bind to this enzyme and inhibit its activity, the bacteria cannot secrete toxins and cause infection in the host.
In conclusion, this study employs computational methodologies, specifically molecular docking and ADMET analysis, to investigate the optimal inhibition values against the target proteins of C. perfringens. Emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside bind to neuraminidase and inhibit its activity; bacteria cannot secrete toxins and do not cause infection in the host. Owing to the experimental validation, the emodin-8-O-β-D-glucopyranoside and physcion-8-O-β-D-glucopyranoside can be defined as drug candidates due to this effect.
References
-
Mehdizadeh Gohari, I.; Navarro, M. A.; Li, J.; Shrestha, A.; Uzal, F.; McClane, B. A. Virulence 2021, 12, 723–753.
[https://doi.org/10.1080/21505594.2021.1886777]

-
Von Itzstein, M.; Wu, W. Y.; Kok, G. B.; Pegg, M. S.; Dyason, J. C.; Jin, B.; Van Phan, T. Nature 1993, 363, 418–423.
[https://doi.org/10.1038/363418a0]
-
Henrissat, B. Biochem. J. 1991, 280, 309–316.
[https://doi.org/10.1042/bj2800309]
-
Kim, J. Y.; Jeong, H. J.; Park, J.-Y.; Kim, Y. M.; Park, S.-J.; Cho, J. K.; Park, K. H.; Ryu, Y. B.; Lee, W. S. Bioorg. Med. Chem., 2012, 20, 1740–1748.
[https://doi.org/10.1016/j.bmc.2012.01.011]
-
Matrosovich, M. N.; Matrosovich, T. Y.; Gray, T.; Roberts, N. A.; Klenk, H.-D. Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 4620–4624.
[https://doi.org/10.1073/pnas.0308001101]
-
Steenbergen, S. M.; Lichtensteiger, C. A.; Caughlan, R. Garfinkle, J.; Fuller, T. E.; Vimr, E. R. Infect. Immun. 73, 1284–1294.
[https://doi.org/10.1128/IAI.73.3.1284-1294.2005]
-
Chen, G.-Y.; Chen, X.; King, S.; Cavassani, K. A.; Cheng, J.; Zheng, X.; Cao, H.; Yu, H.; Qu, J.; Fang, D.; Wu, W.; Bai, X.-F.; Liu, J.-Q.; Woodiga, S. A.; Chen, C.; Sun, L.; Hogaboam, C. M.; Kunkel, S. L.; Zheng, P.; Liu, Y. Nat. Biotechnol. 2011, 29, 428–435
[https://doi.org/10.1038/nbt.1846]
-
Wang, Y.-H. Front. Cell. Infect. Microbiol. 2020, 9, 462.
[https://doi.org/10.3389/fcimb.2019.00462]
-
Van Immerseel, F.; De Buck, J.; Pasmans, F.; Huyghebaert, G.; Haesebrouck, F.; Ducatelle, R. Avian Pathol. 2004, 33, 537–549.
[https://doi.org/10.1080/03079450400013162]
-
Soong, G.; Muir, A.; Gomez, M. I.; Waks, J.; Reddy, B.; Planet, P.; Singh, P. K.; Kaneko, Y.; Wolfgang, M. C.; Hsiao, Y.-S.; Tong, L.; Prince, A. J. Clin. Invest. 2006, 116, 2297–2305.
[https://doi.org/10.1172/JCI27920C1]
-
Khalil, A. A. K.; Park, W. S.; Kim, H. J.; Akter, K. M.; Ahn, M.-J. Nat. Prod. Sci. 2016, 22, 220–224.
[https://doi.org/10.20307/nps.2016.22.3.220]
-
Khalil, A. A. K.; Park, W. S.; Lee, J.; Kim, H.-J.; Akter, K.-M.; Goo, Y.-M.; Bae, J.-Y.; Chun, M.-S.; Kim, J.-H.; Ahn, M.-J. Arch. Pharm. Res. 2019, 42, 505–511.
[https://doi.org/10.1007/s12272-019-01160-x]
-
Lachowicz, S.; Oszmiański, J. Molecules 2019, 24, 1436.
[https://doi.org/10.3390/molecules24071436]
-
Zeng, H.; Wang, Y.; Gu, Y.; Wang, J.; Zhang, H.; Gao, H.; Jin Q.; Zhao L. Life Sci. 2019, 218, 25–30.
[https://doi.org/10.1016/j.lfs.2018.08.013]
-
Lin, C.-J.; Lin, H.-J.; Chen, T.-H.; Hsu, Y.-A.; Liu, C.-S.; Hwang, G.-Y.; Wan, L. PLoS One 2015, 10, e0117602.
[https://doi.org/10.1371/journal.pone.0117602]
-
Su, P.-W.; Yang, C.-H.; Yang, J.-F.; Su, P.-Y.; Chuang, L.-Y. Molecules 2015, 20, 11119–11130.
[https://doi.org/10.3390/molecules200611119]
-
Liu, F.; Li, F.-S.; Feng, Z.-M.; Yang, Y.-N.; Jiang, J.-S.; Li, L.; Zhang, P.-C. Phytochemistry 2015, 110, 150–159.
[https://doi.org/10.1016/j.phytochem.2014.12.007]
-
Uddin, Z.; Song, Y. H.; Curtis-Long, M. J.; Kim, J. Y.; Yuk, H. J.; Park, K. H. 2016, 193, 283–292.
[https://doi.org/10.1016/j.jep.2016.08.026]
-
Ke, J.; Li, M.-T.; Xu, S.; Ma, J.; Liu, M.-Y.; Han, Y. Pharm. Biol. 2023, 61, 177–188.
[https://doi.org/10.1080/13880209.2022.2158349]
-
Gill, S. C.; Von Hippel, P. H. Anal. Biochem. 1989, 182, 319–326.
[https://doi.org/10.1016/0003-2697(89)90602-7]
-
King, A. D.; Pržulj, N.; Jurisica, I. Methods Mol. Biol. 2012, 2012, 297–312.
[https://doi.org/10.1007/978-1-61779-361-5_16]
- Biovia D. S. Discovery studio modeling environment. San Diego, Dassault Systemes, Release, 2015, p 4.
-
Dundas, J.; Ouyang, Z.; Tseng, J.; Binkowski, A.; Turpaz, Y.; Liang, J. Nucleic Acids Res. 2006, 34, W116–W118.
[https://doi.org/10.1093/nar/gkl282]
-
Wallace, A. C.; Laskowski, R. A.; Thornton, J. M. Protein Eng. 1995, 8, 127–134.
[https://doi.org/10.1093/protein/8.2.127]
-
Trott, O.; Olson, A. J. J. Comput. Chem. 2010, 31, 455–461.
[https://doi.org/10.1002/jcc.21334]
-
Lee, Y.; Youn, H.-S.; Lee, J.-G.; An, J. Y.; Park, K. R; Kang, J. Y.; Ryu, Y. B.; Jin, M. S.; Park, K. H. Eom, S. H. Biochem. Biophys. Res. Commun. 2017, 486, 470–475.
[https://doi.org/10.1016/j.bbrc.2017.03.064]
-
Laskowski R. A,; Swindells M. B. LigPlot+: multiple ligand-protein interaction diagrams for drug discovery. ACS Publications, Washington, DC, 2011, pp 2776–2786.
[https://doi.org/10.1021/ci200227u]
- Center for Disease Control. Antibiotic Resistance Threats in the United States; U.S. Department of Health and Human Services; Washington DC, USA, 2013, 1–114.
- O’Neill, J. Tackling Drug-Resistant Infections Globally: Final report and recommendations; Wellcome Trust & HM Government, London, UK 2016, pp 1–84
-
Wencewicz, T. A. J. Mol. Biol. 2019, 431, 3370–3399.
[https://doi.org/10.1016/j.jmb.2019.06.033]
-
Fatima, M.; Amin, A.; Alharbi, M.; Ishtiaq, S.; Sajjad, W.; Ahmad, F.; Ahmad, S.; Hanif, F.; Faheem, M.; Khalil, A. A. K. Molecules 2023, 28, 2635.
[https://doi.org/10.3390/molecules28062635]